Atomistic Simulations
Atomistic simulations model materials at the level of atoms and are broadly employed to gain insight in their behavior. Density Functional Theory (DFT) is computational quantum mechanical method used to foresee the properties of molecules and bulk materials. It is a technique for investigating the electronic structure (principally the ground state) of many-body systems. DFT simulation can predict a vast range of structural, chemical, optical, spectroscopic, elastic, vibrational and thermodynamic phenomena. Molecular dynamics (MD) instead is a simulation method used to investigate the dynamic evolution of a system. The trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion, where forces between the particles and their potential energies are often calculated using known interatomic potentials or molecular mechanics force fields.
Recently we employed state-of-the-art computational codes for the realization of the following projects.
Design of novel catalysts for CO2 and O2 electroreduction
The aim of this project is design and optimization of novel catalysts for O2 and CO2 electroreduction to desired products with high activity, selectivity and stability by means of a computational approach based on DFT calculations.
In particular, we predict and tailor the structural and electrocatalytic properties of bimetallic Cu/M alloys towards efficient and selective CO2RR and of doped/decorated reduced graphene oxide towards efficient and selective CO2RR and ORR [1]. Theoretical understanding of the reaction mechanisms and of the intermediate steps of reduction at the electrocatalyst’s surfaces guides the synthesis of materials and allows a deeper comprehension of the catalytic process.
- N. Garino, M. Castellino, A. Sacco, F. Risplendi, J. A. Muñoz-Tabaresa, M. Armandi, A. Chiodoni, M. Quaglio, C. F. Pirri and G. Cicero, ”Mn-N co-doping of reduced graphene oxide via microwave-assisted route: a novel electrocatalyst for the oxygen reduction reaction” submitted;
Graphene based selective membranes for desalination and gas separation
To reduce production costs of filtration membranes, carbon-based filters have been largely investigated especially focusing on the design of optimal nanoporous membranes showing high water permeability and excellent selectivity with respect to a specific solute. The aim of this research activity is to develop a new generation of reverse osmosis (RO) membranes based on graphene materials (such as graphene and graphene oxide) able to reduce contaminants concentrations in the permeate employing a theoretical approach based on density functional theory (DFT) and classical molecular dynamics (MD). Computer simulations have been employed first to examine the effects of the oxidation of graphene on its structure and electronic properties. In particular we aimed at finding correlations between the composition of GO, its structural characteristics and electronic properties [2]. Subsequently, we studied how GO structure determines the size of pores generated in GO upon thermal treatment.
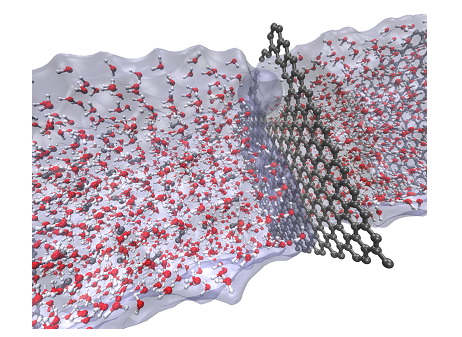
Representation of the filtration model system.
Finally, we have also investigated the relationship between the atomic structure of nanoporous graphene and its membrane properties in RO applications by means of classical MD simulations. We calculated for different pore sizes water permeability and identify the optimal pore size for RO applications. We also established the implications of graphene-based materials as promising membrane for boron removal during RO filtration [3].
In a complementary study, we also focused on understanding the features that the graphene pores must possess to accomplish CO2/CH4 and H2/CH4 separation during natural gas. By means of MD simulations we demonstrated how pore shape, dimension and chemical functionalization determine the graphene membrane permeance and selectivity [4].
2. F. Savazzi, F. Risplendi, G. Mallia, N. M. Harrison and G. Cicero, “Unravelling Some of the Structure-Property Relationships in Graphene Oxide at Low Degree of Oxidation “ J. Phys. Chem. Lett. 9, 1746-1749 (2018);
3. F. Risplendi, F. Raffone, L.-C. Li, J. C. Grossman and G. Cicero, “Molecular Simulation of Solvation Properties of Boric Acid in Water Solution and their Separation by means of Reverse Osmosis Membrane” submitted;
4. G. Tronci, F. Raffone and G. Cicero, “Theoretical Study of Nanoporous Graphene Membranes for Natural Gas Purification”, Appl. Sci. 8, 1547 (2018);
Solid/water interfaces
This project regards the study of interfaces between a solid material and water by means of ab initio simulations based on ab initio MD. Initially we studied the behavior of cubic Silicon Carbide surfaces (a biocompatible material) exposed to water molecules. This study highlighted that the interaction of SiC with water depends on the surface termination of this material: the C-terminated surfaces show a hydrophobic behavior while the Si-terminated surfaces show a hydrophilic behavior. In other words, a simple material like SiC can show a very different macroscopic behavior, when interacting with water, engineering the atomic termination of its surface [5]. This is surely an interesting finding not common to many materials.

Ball-and-stick rendering of three representative configurations explored during ab initio simulations of water deposition on the SiC(001) surface and of the resulting solid/liquid interface.
We also showed that water, close to a surface, is characterized by molecular density, which is larger than the density of pure bulk water [6]. Moreover, the water molecules at the interface present dipole moments which are different from bulk water, correspondingly also the electronic properties and the IR spectrum of surface water show markedly different features [7]. In addition, we were interested in understanding how water wets a gold surface and also to understand how protein/gold interaction is mediated by water. In particular we showed that water molecules in contact with gold surfaces strongly modify their ability to form hydrogen bonds [8,9] and that these water molecules play a determining role in mediating the interaction of the gold surface with the beta sheet of a protein.
5. G. Cicero, A. Catellani and G. Galli ,“Atomic control of water interaction with biocompatible surfaces: The case of SiC(001)”, Phys. Rev. Lett. 93, 016102/1-4 (2004);
6. G. Cicero, J. C. Grossman, A. Catellani, G. Galli, “Water at hydrophilic solid surface probed by ab initio molecular dynamics: inhomogeneous thin layers of dense fluid.” J. Am. Chem. Soc. 127, 6830 (2005)
7. D. Donadio, G. Cicero, E. Schwegler, M. Sharma and G. Galli, “Electronic effects in the IR spectrum of water under confinement”, J. Phys. Chem. B 113, 4170 (2009);
8. A. Calzolari, G. Cicero, C. Cavazzoni, R. Di Felice, A. Catellani and S. Corni, “Hydroxyl-Rich β-Sheet Adhesion to the Gold Surface in Water by First-Principle Simulations” J. Am. Chem. Soc. 132, 4790 (2010);
9. G. Cicero, A. Calzolari, S. Corni, and A. Catellani, “Anomalous wetting layer at the Au(111) surface”, J. Phys. Chem. Lett. 2, 2582 (2011).
Chemo-physical properties of ZnO Nanowires
ZnO nanostructures find important applications in the field of mechanical energy harvesting devices (piezotronics), in gas sensors, in optoelectronic and memristive devices.
Regarding nano-mechanical energy harvesting we gave an important contribution developing a novel method to calculate the piezoelectric response of nanostructures by means ab initio DFT calculation [10] separating the response of the structure into bulk and surface contributions. This novel approach has been later applied to predict the piezoelectric properties of ZnO nanowires [11].
We also unveiled the gas sensing mechanism of ZnO nanowire [12]: for the first time an ab initio study has proven that ethanol sensing by ZnO nanowires in mediated by the oxygen molecules present in the atmosphere in normal conditions and pre-adsorbed at the nanowire surfaces. This finding has important consequences for the optimization and the increase of the sensor sensitivity and selectivity.

Side view of the optimized O2/ZnO(11̅00) interface (a) and ethanol/ZnO(11̅00) interface (b) represented along the [12̅10] (left structures) and [0001̅] (right structures) directions
A combined theoretical and experimental investigation of the optoelectronic properties of ZnO nanostructures is reported in paper [13]. The ab initio calculations were fundamental to understand the role of Zn vacancies at surface in generating green luminescence in ZnO nanostructures.
G. Cicero’s group has been recently involved also in the study of memristive devices. We unveiled the switching mechanism between high and low resistive state of ZnO NWs-based memristors via DFT calculations, by studying the extraction and diffusion of copper metal atoms from the electrode to the ZnO NWs surface. On the basis of the theoretical results, it has been proposed a novel switching mechanism relying on ZnO surface doping effects of single Cu adatoms rather than on the formation of a continuous metallic filament at variance with other studies where current has been considered a result of the electron flow through metallic nanoparticles (NPs) separated by nanometric gaps [14]. Further, in paper [15] we demonstrate theoretically that core-shell structures based on polyacrylic acid coated ZnO nanowires exhibit a resistive switching behavior characterized by internal multiple resistance states, owing to the changes in surface states induced by redox reactions occurring at their surfaces. The mechanism of switching between multiple steps was associated with redox reactions involving species at the interface (e.g. methanal or hydroxyl groups).
10. G. Cicero, A. Ferretti, and A. Catellani, ”Surface-induced polarity inversion in ZnO nanowires”, Phys. Rev. B 80, 201304 (2009);
11. K. K. Korir, G. Cicero and A. Catellani, “Piezoelectric properties of zinc oxide nanowires: an ab initio study” Nanotechnology 24, 475401 (2013);
12. K. K. Korir, A. Catellani and G. Cicero “Ethanol gas sensing mechanism in ZnO nanowires: An ab initio study” Journal of Phys. Chem. C 118, 24533-24537 (2014);
13. F. Fabbri, M. Villani, A. Catellani, A. Calzolari, G. Cicero, D. Calestani, G. Calestani, A. Zappettini, B. Dierre, T. Sekiguchi and G. Salviati “Zn vacancy induced green luminescence on non-polar surfaces in ZnO nanostructures" Scientific Reports 4, 5158 (2014);
14. F. Raffone, F. Risplendi, and G. Cicero “A New Theoretical Insight into ZnO NWs Memristive Behavior” Nano lett. 16, 2543-2547, (2016);
15. S. Porro, F. Risplendi, G. Cicero, K. Bejtka, G. Milano, P. Rivolo, A. Jasmin, A. Chiolerio, C. F. Pirri, and C. Ricciardi, “Multiple resistive switching in core-shell ZnO nanowires exhibiting tunable surface states”, J. Mat. Chem. C 5, 10517-10523 (2017).
Functionalization of semiconductor surfaces with organic molecules
We applied ab initio methods to understand how organic molecules can be chemically attached to several type of semiconductor surfaces [e.g SiC Ref. 16] and to understand how the mechanical properties of surfaces are affected by an organic layer covalently attached to the surface [17-18]. In particular the studies presented in [17] and [18] have highlighted the importance of the surface stress and of the variation of the surface elastic constants induced by surface functionalization during mechanical detection.

(a) Structure of methylthiolate adsorbed on Au(111) (top view) (b) Side view of the same system indicating isosurfaces of the charge-density difference upon adsorption.
16. G. Cicero and A. Catellani, “Towards SiC surface functionalization: an ab initio study.”, J. Chem. Phys. 122, 214716 (2005);
17. F. Risplendi, A. Ricci and G. Cicero, “Functionalization layer effect on the mechanical properties of silicon based micro-cantilever mass sensors: A theoretical study”, Sensors and Actuators B: Chemical 195, 177 (2014);
18. V. Scrinivasan, G. Cicero and J. C. Grossman, “Adsorption-Induced Surface Stresses in Alkanethiolate-Au Self-Assembled Monolayers”, Phys. Rev. Lett. 101, 185504 (2008).
Dye sensitized oxides films in third generation solar cells
In third generation solar cells, surface effects are very important and may be fundamental in determining the efficiency of the device. The aim of this study was to understand the role of the anchoring group of the dye in affecting the efficiency of dye sensitized solar cells (DSCs) [19-20] and of the influence of the co-adsorbent in determining the level alignment of the heterostructure [21]. This work aimed at understanding the electronic and optical properties of the interface between TiO2 or ZnO and an innovative organic dye (hemi-squaraine). Hemi-squaraine presents a functional group called squaric acid, that we amployed for the first time in Dye Sensitized Cells (DSC). Starting from the observation that the aromaticity of the squaric acid moiety allows to extend the electron delocalization of the benzoindolic group of the dye toward the surface of the oxide substrate, we evaluate the possibility of employing this dye and to understand the mechanisms that rule the electron transfer at the oxide/hemi-squaraine interface and determine the conversion efficiency of the solar cell. We performed ab initio simulations and found very strong interaction of the squaric acid with the anatase surface. The chemical bonds with this oxide are stronger than that formed by other dye anchoring groups such as the isonicotinic acid or the cathecolic acid often employed in DSC.

Ball-and-stick representation of CT1 attached to the anatase-TiO2(101) surface. The electron density isosurface of the interface states involving CT1’s HOMO (red isosurface) as well as LUMO and LUMO+1 (blue isosurface) are also reported.
The analysis of the electronic structure of the hybrid organic/inorganic interface has highlighted that the system realizes a type II heterostructure, which favors the adiabatic electron transfer of the electron from the dye molecule to the oxide.
19. G. Cicero, G. Musso, A. Lamberti, B. Camino, S. Bianco, D. Pugliese, F. Risplendi, A. Sacco, N. Shahzad, A. M. Ferrari, B. Ballarin, C. Barolo, E. Tresso and G. Caputo “Combined experimental and theoretical investigation of the hemi-squaraine/TiO2 interface for dye sensitized solar cells”, Phys. Chem. Chem. Phys. 15, 7198 (2013);
20. N. Shahzad, F. Risplendi, D. Pugliese, S. Bianco, A. Sacco, A. Lamberti, R. Gazia, E. Tresso and G. Cicero “Comparison of Hemi-Squaraine Sensitized TiO2 and ZnO Photoanodes for DSSC Applications”J. Phys. Chem. C 117, 22778 (2013);
21. F. Risplendi and G. Cicero, “Co-Adsorbent Effect on the Sensitization of TiO2 and ZnO Surfaces: A Theoretical Study”J. Phys. Chem. C 119, 27348 (2015).
Study of inorganic semiconductors and of their interfaces
InN is a low band gap material proposed as an innovative material to be employed in the form of nanowires in third generation solar cells and optoelectronic devices. The results achieved by our group and presented in papers [22,23] mainly regard: understanding the fundamental electronic properties of InN nanowires [22], understanding the role of native indium oxide (In2O3) on the properties of InN nanowires [22-24]. Other important results were achieved in the field of InN Nanowires such as: understanding the growth mechanism of InN Nanowires [25], understanding the role of the nitrogen vacancies in determining the intrinsic n-type behavior of InN nanowires [26] and devising an organic functionalization protocol for InN nanowires [27].

Contour plot of the PES for In adatom adsorption on repeated 2 × 2 cells on the m-plane (a) and a-plane (b) surfaces.
In the last few years, our group has also started working on 2D semiconductors belonging to the class of transition metal dichalcogenides, such as MoS2. We demonstrated for the first time how it is possible to tailor the phase stability of MoS2 by alloying it with SnS2 [28].
22. A. Terentjevs, A. Catellani, D. Prendergast and G. Cicero “Importance of on-site corrections to the electronic and structural properties of InN in crystalline solid, nonpolar surface, and nanowire forms”, Phys. Rev. B 82, 165307 (2010);
23. A. Aliano, A. Catellani and G. Cicero, “Characterization of amorphous In2O3: an ab initio Molecular Dynamics study”, Appl. Phys. Lett. 99, 211913 (2011);
24. A. Aliano, G. Cicero and A. Catellani “Origin of the accumulation layer at the InN/a-In2O3 interface”, ACS Applied Materials and Interfaces 7, 5415 (2015);
25. A. Aliano, A. Catellani and G. Cicero, “Nitrogen vacancies at InN (1-100) surfaces: A theoretical study”, Appl. Phys. Lett. 96, 171901 (2010);
26. A. Molina-Sánchez, A. García-Cristóbal, A. Cantarero, A. Terentjevs and G. Cicero “LDA+U and tight-binding electronic structure of InN nanowires”, Phys. Rev. B 82, 165324 (2010);
27. A. Aliano, A. Catellani and G. Cicero, “Adatom kinetics on nonpolar InN surfaces: Implications fro one-dimensional nanostructures growth” Appl. Phys. Lett. 99, 193106 (2011);
28. F. Raffone, C. Ataca, J. C. Grossman and G. Cicero “MoS2 Enhanced T-Phase Stabilization and Tunability Through Alloying”, J. Phys. Chem. Lett. 7, 2304–2309 (2016).